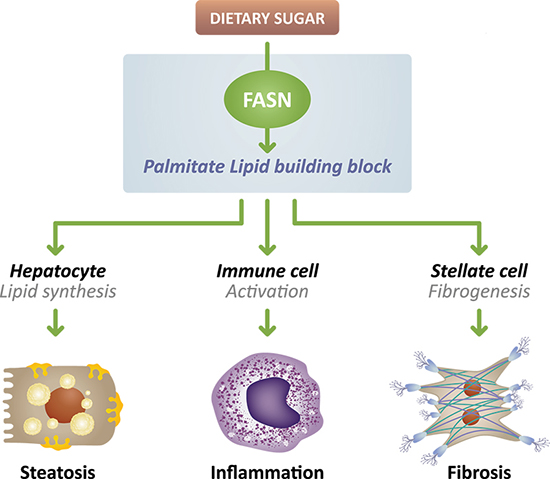
The critical role of FASN overactivity in metabolic dysfunction-associated steatohepatitis (MASH), formerly known as nonalcoholic steatohepatitis (NASH), acne and certain cancers has made it an attractive target for drug therapy. FASN is a key enzyme in de novo lipogenesis (DNL) pathway that converts metabolized of dietary sugars such as fructose into a saturated fatty acid, palmitate. This saturated fatty acid building block is used to make longer chain, polyunsaturated fatty acids used by the cell for energy production, and itself is an important component of cellular signal transduction pathways.
In diseases such as MASH, acne and in certain cancers, this pathway does not function properly and overproduces lipids—causing unfavorable metabolic and inflammatory problems to occur.
In MASH, this promotes excess accumulation of fat in the liver, which can damage the liver and stimulate inflammation and fibrotic scarring, thus targeting FASN overactivity may provide the opportunity to simultaneously tackle several components of disease.
In acne, FASN is responsible through lipid synthesis for the production of skin oils (sebum). More than 80% of key sebum lipids such as palmitate and sapienic acid are produced by DNL/FASN. In acne, excess sebum can lead to skin lesions and is a pro-inflammatory stimulus leading to exacerbation of those lesions, including development of nodules (nodular acne) and cysts (cystic acne).
Denifanstat impacts key drivers of MASH
Following a rigorous medicinal chemistry and preclinical research effort, we selected denifanstat from our library of more than 1,200 internally discovered and wholly owned FASN inhibitors. Denifanstat is designed to bind to the FASN protein in an area that is not an enzymatic active site and unique to the structure of FASN. This selectivity is critical for preventing off-target effects that plagued earlier generations of FASN inhibitors. We advanced denifanstat into clinical development based on its oral administration, high selectivity for FASN, and excellent pharmacokinetic and pharmaceutical properties, including restricted penetration of the blood-brain barrier.
Our preclinical data published in Nature demonstrated that FASN inhibition targets multiple drivers of NASH by reducing steatosis, inflammation and fibrosis in preclinical models of NASH. In another study conducted at the Icahn School of Medicine at Mt. Sinai Hospital in New York, mice with NASH that progressed to develop hepatocellular carcinoma were treated with a FASN inhibitor that not only reversed fibrosis but also reduced the number of liver tumors compared to animals that received placebo. Several treated animals were tumor-free at the end of the study.
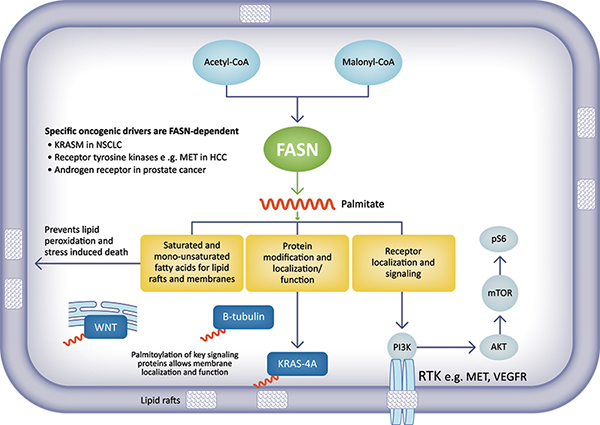
In preclinical cancer models, our FASN inhibitors have demonstrated significant anti-tumor effects, especially in tumor cells that are dependent on FASN for their survival, including non-small cell lung cancer with Kirsten rat sarcoma (KRAS) mutations, and certain metastatic-resistant prostate cancers and hepatocellular carcinomas.
Our extensive compound library of FASN inhibitors provides us the ability to evaluate additional drug candidates for further development as warranted.
FASN plays a key role in molecular mechanisms associated with cancer